At MacResearch we have some big plans for Xgrid in the coming months. Stay tuned for announcements. But to warm you up, we have a real coup: Charles Parnot, creator of one of the largest Xgrids on the planet, has agreed to write a series of tutorials for MacResearch. To kick off the series, Charles covers basic command line use. [MR]
You have a few thousands Xserve in your basement and you don’t know what to do with them. Or more realistically, there are a bunch of macs in your lab, old and new, big and small, desktop and laptops, idle all night and most of the day. And you are thinking: “What a waste! All the things we could do with these machines!”. Well, waste no more! As you may know, Apple has the solution for you, they call it Xgrid.
In this tutorial, I will try to guide you through all the steps necessary to get something “real” running with Xgrid. I decided to use as an example Fasta, a program used by biologists to identify DNA sequences. This is quite a good fit for Xgrid: relatively little data, long computations, easily parallelizable. I plan to have at least 3 installements for this tutorial, of increasing complexity. This first installment is aimed at scientists not necessarily familiar with Xgrid or with parallel computation, and will include a quick introduction to the command-line environment. Hopefully, it won’t be too boring for scientists already very familiar with some of these concepts, but eager to learn more about Xgrid too.
Think Xgrid
Before learning how to use Xgrid, you should determine if it even makes sense to use it and if you will actually be able to take advantage of it. On the xgrid-users mailing list, many ask if Xgrid can boost applications like iMovie, Adobe Photoshop or Final Cut Pro. The answer, in short, is “No”. Unfortunately, Xgrid is not yet the silver bullet that can magically take an existing program such as iMovie, cut it in slices and run in on all the macs it can find in the neighborhood. The “cut in slices” part is still your job, or in the case of iMovie, it would be iMovie’s job. It is difficult to give a simple answer to the simple question “Can Xgrid help to run my calculations?”. It depends not only on the type of tasks you want to run but also on the scale of the project (how many tasks?). In this section, I will show you how to distinguish between what Xgrid can and can’t do.
Let’s start with a non-computational analogy, with something that scientists do a lot: writing papers. If you work alone on on a paper, maybe it will take 1 week to write 10 pages (working really hard!). You could ask a graduate student to write it, but that would probably take 2 weeks. So here is the solution: graduate students are cheap, just get 10 graduate students to work on it, one page each, it will take 2 weeks / 10 = 1.4 days. Problem solved! Problem solved?… Well, I hope you have realized 2 things: (1) you should have more respect for graduate students; (2) it won’t work. Writing papers is not amenable to parallelization. Xgrid is not good at the computational equivalent of writing papers, which requires tight integration between the different parts of the job [1].
Next, imagine you are a world-wide expert on nicotinic receptors and you have to write a 150 page review. Before writing it, you need the full-text versions of all the relevant publications on the subject, but this is going to take ages because there are 800 of them, buried in various journals scattered throughout the library. Well, the graduate students can help! If that task takes 2 weeks for one person, it might take just 1 or 2 days if 10 graduate students work on it. Here, I hope you have realized 2 things: (1) you should still have more respect for graduate students, (2) it will work (maybe by adding free pizza too). Making photocopies of articles at the library is amenable to parallelization. Xgrid is very good at the computational equivalent of sending a horde of graduate students in a library.
If we go back to “real” computational tasks, here are some examples of calculations where Xgrid can help, because they can be broken down in small independent tasks that can each be computed in separate machines, in parallel:
- Animation movies (think Pixar!), where each task is the calculation for just one frame of the movie (the image corresponding to 1/24th of a second)
- Drawing Mandelbrot fractals, where each task is a small piece of the full fractal
- Analyzing radio signal from space for signs of intelligence, where each task deals with the signal from a small amount of time or a small area of the sky (or both), also known as SETI@home
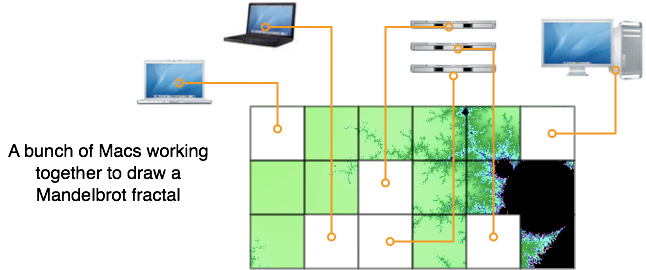
Xgrid will be best at what are called “embarrassingly parallel” problems. You have to be able to divide your computation in dozens or hundreds of smaller tasks, where each task would take a few minutes to a few hours on one processor [2].
Here are now some examples of calculations that are not Xgrid-friendly:
- Simulating the molecular movements of a protein, because you can only calculate the next step when the previous step is finished
- Calculating the Fibonacci series, because each new value is dependent on the previous ones: u(n+2)=u(n+1)+u(n)
- Compress an image into jpeg format, because it requires to process information about the whole image
However, even in the case of non-parallelizable tasks like above, Xgrid can be useful if you need to perform the same tasks over and over again with different conditions. For instance, to model the molecular dynamics of a protein (first example above), it is quite frequent to run several simulations with variable starting positions. This allows to explore the conformational space of the protein (see also [3]). It is trivial to run several independent simulations, one on each machine, a configuration where Xgrid can be useful. Similarly, while compressing one image might be hard to parallelize, compressing hundreds of images is easy to do in parallel on several machines. These examples show that Xgrid can be useful even for tasks that are not parallelizable to begin with. The whole project itself might be amenable to parallelization because you want to run many of these tasks.
One more thing… Before you can run a task on a computer, you need to start by sending some files: the program itself, but also the data to be processed. When the task is done, the results needs to be sent back too. In the example of image compressing, the compression algorithm is usually relatively fast. If transfering the image files back and forth takes as much time as running the program itself, you have not gained anything. This additional time actually needed to run a job, and wasted on data transfer, is also called “network overhead”. The network overhead is smaller for molecular dynamics simulation: the amount of data needed to describe a protein structure is relatively small, while the simulation itself is processor-intensive and takes a while.
In conclusion, Xgrid will be most useful if:
- The calculations are naturally amenable to parallelization (e.g. fractals)
- And/or you have to repeat the same calculations many times (e.g. animation movies)
- The network overhead is not too big
In all cases, you will need at some point to have a good understanding of the computations you want to perform and of the programs you want to run with Xgrid. There is no way around that.
Start Xgrid
I won’t spend too much time explaining the Xgrid architecture or how to get Xgrid running, as there is a wonderful tutorial to get you started. I will try to cover the basics here so you can get started quickly. The Xgrid terminology is very simple:
- the “client” sends jobs to the “controller”
- the “controller” queues the jobs and sends them to the “agent”
- the “agent” runs the job
Of course, the whole point of Xgrid is to have several agents, all working in parallel, each on a different job. Similarly, there can be several clients. For instance several members of your lab could submit jobs, each from their own computer. And this is basically all there is to understand about Xgrid! Now, let’s get these different pieces in place on your machine.

If you have already an Xgrid controller running with agents connected, you don’t need my help here. You can just skip to the next section. But if you have never used Xgrid before, I want to make the setup as simple as possible, so you can run the tutorial now and build a full-fledge Xgrid solution later. To get started immediately, download this package. After unzipping, you should have a folder with 3 applications in it: ‘xgrid-start’,’xgrid-stop’ and ‘xgrid-clean’.
All 3 will behave the same when double-clicked: a window will open in Terminal and you will need to enter your administrator password before the automagical script gets executed.
The first application ‘xgrid-start’ will start a controller and an agent on the machine you are using. The second application ‘xgrid-stop’ will stop them. For security reason, it is highly recommanded to run ‘xgrid-stop’ when done with the tutorial. The third one ‘xgrid-clean’ will give you a fresh start, with a brand new controller that don’t remember any of the previous jobs or agents it has seen (some people like to clean things).
To follow this tutorial, simply run ‘xgrid-start’ and you can then use your machine as the client, controller and agent, all three at the same time! It works just fine for testing purposes, even if it sounds a bit silly: your computer submits a job to itself, then it decides to assign that job to itself, because there is no one else around.
Finally, to check visually how things are going, I strongly recommand you install the free Xgrid Admin program developed by Apple. It will display all the agents and all the jobs attached to your controller. It is very easy to use, so I will let you play with it on your own.
After you have a controller and an agent running, you can submit jobs as a “client”. As you realize, the client is the most interesting and creative part: you, the scientist! The rest of this tutorial will be all about understanding the client.
Your friend the Terminal
Before we talk more about Xgrid, we need to talk about the Terminal and the “command-line environment”. For some, it might be scary, and if you are afraid of typing words instead of clicking buttons, now is the time to overcome that fear. There is nothing really hard about the command-line. All you do is type a command and the computer executes that command. When it is done, the computer patiently waits until you enter the next command.
To get started, double-click the application “Terminal” (in /Applications/Utilities). A new window should open that welcomes you with this very interesting message (or something similar):
mycomputer:~ jdoe$
This is called the “prompt”, and it lets you know that the Terminal is waiting for you to type a command (for clarity, I won’t include the prompt in the examples). So let’s type a command! Drop your mouse, grab your keyboard, and try this (hit return to validate):
ls /Applications/Utilities
The Terminal will understand that you want to “list” the contents of your Utilities folder (“ls” is a shortcut for “list” because, you know, a four-letter word, this is too much typing). When execution of the command is finished, you get the prompt again. Next, you could abuse your new power and order:
say "hello"
I hope you find that last one fun! OK, I admit this is quite useless (somebody in the back said “pathetic”?). But the Terminal can in fact be extremely powerful (and the more curious may want more details). Every time you type a command, you actually start a program on your machine, just like you would start iTunes or Excel. Except the program is invisible! It runs in the background, without taking any space on your screen. The command we typed above did not take very long to complete: the program “say” was only running for a few instants. However, some other invisible program could run long and complicated calculations, save the results to disk and send you an email when done. You could even log out of the computer and the invisible program would keep running.
I hope you are now very excited about those faceless programs… You’d better be, because they are the only programs that Xgrid can run. You cannot ask Xgrid to open Safari or iMovie. Xgrid will only understand “ls /Applications/Utilities” or other more interesting commands.
Your buddy Fasta
Now that you are all excited about the Terminal, let me introduce the program that we will use with Xgrid. The name is “Fasta” [4]. The main purpose of Fasta is to identify in a large collection of sequences the pieces that are similar to a given sequence. You might want to think of Fasta as Google for DNA. For instance, you may have just found a mutation that makes worms able to solve second-degree equations. Then you wonder if there is a similar piece of DNA in humans. What do you do? You Google it? Errr… Nooo… You Fasta it! You run Fasta using the piece of worm DNA as the “query”, and the human genome as the “library”, and boom! You find the human gene for second-degree equation solving.
But before starting a revolution in genetics, let’s install all we need for this Xgrid/Fasta tutorial. I have prepared a little package for you. After downloading, move it to the folder called “Shared” that you will find in the “Users” folder [5], then double-click on it to extract all the files. You should now have a folder called “fasta-tutorial” that contains the “fasta” program (lowercase ‘f’), as well as all the human chromosomes [6] and several test sequences. The reason we put these files in this Shared folder is that Xgrid only has access to a limited number of places in the system, for security reasons, and “Shared” is one of them. If you have several agents, you will need to repeat that installation for each one of them (in a next installement, we will explore alternatives to such a tedious process; see also [7]).
Update: with Leopard 10.5, access to the filesystem has been considerably restricted, and the xgrid agent now runs in a ‘sandbox’, which reduces access to certain folders, including /Users/Shared. The tutorial would need to be updated to take that into account, for instance by using /tmp instead. In the meantime, sorry about the trouble!
We are now ready to type our first fasta command using our friend the Terminal:
fasta magic-worm-gene.seq chromosomeY.fa
What this does is ask the computer to run the program “fasta” using “magic-worm-gene.seq” as the query, and “chromosomeY.fa” as the reference library [8]. In other words, look for the magic-worm-gene sequence in the Y chromosome. However, the above command returns an error message… Terminal, you are not my friend anymore! What happens is your computer is really dumb (not you). Even though you just put these files on your hard drive, and it should be obvious that the next thing you do is use these files, you still have to tell the Terminal where those files are. Instead of just saying “fasta” you should say “/Users/Shared/fasta-tutorial/fasta” so that your dumb computer knows where to look. The command you should write is then much longer, so make sure all the following is typed as one continous line with no carriage returns (it may appear as 2 lines on your web browser and in the Terminal):
/Users/Shared/fasta-tutorial/fasta \
-q /Users/Shared/fasta-tutorial/magic-worm-gene.seq /Users/Shared/fasta-tutorial/chromosomeY.fa
In this command, I also added a “-q”, which will prevent fasta from asking additional information that is irrelevant for this tutorial (see documentation via ftp). Because the command is quite long, it has been spread over two lines, and the ‘\’ continuation character used, but you can also write it all on one line if you choose (without the ‘\’, of course). If all is right, after you hit return, you first get some message that describes the query. Then the program starts scanning the Y chromosome and searching for something that looks like the piece of DNA you provided. While this happens, you do not get the prompt, and nothing happens on the screen. But your invisible buddy Fasta is running! When the calculation is actually finished, you get a bunch of interesting lines on your terminal display, that show you all the sequences similar to the query “magic-worm-gene.seq”, and then finally the prompt. You buddy Fasta is done, and your friend the Terminal is waiting for the next command.
Use Xgrid
If you look at the result above, the gene was not found in the Y chromosome, so we have to scan more of the human genome. It took several seconds to scan the Y chromosome, but this is the smallest chromosome (yet it makes half of the world quite different from the other half…). There are 22 more, and then the X chromosome. Scanning all of these would probably take a while. What if you wanted to also scan the rat genome, the worm genome, the rice genome and the drosophila genome? Or every sequence known out there? And what if you have more than one query? Maybe you want to compare every single worm gene to all the other genomes? This is going to take a while even on a Mac Pro Dual processor 3.0GHz Dual-Core Intel Xeon. What would you do if you had instead a room full of computers? To run the above query on every human chromosome, you could sit 24 graduate students in front of 24 computers, tell them to open the Terminal and have them type a different query on each machine:
machine1: fasta magic-worm-gene.seq chromosomeX.fa
machine2: fasta magic-worm-gene.seq chromosomeY.fa
machine3: fasta magic-worm-gene.seq chromosome1.fa
machine4: fasta magic-worm-gene.seq chromosome2.fa
machine5: fasta magic-worm-gene.seq chromosome3.fa
...
machine24: fasta magic-worm-gene.seq chromosome22.fa
As you will see, using Xgrid is very similar to this approach, but without the graduate students. You can just sit alone at your desk and type everything from your own machine. We are now going to act as an Xgrid client, by sending a job to our Xgrid controller. If you have setup your machine using the ‘xgrid-start’ provided with this tutorial, then the controller is actually the same machine as the client. In this case, the address of your controller is simply ‘localhost’ (it is the computer way of saying ‘me’ when talking about itself). In the Terminal, we will first tell xgrid about that:
export XGRID_CONTROLLER_HOSTNAME=localhost
Then, to submit the chromosome Y job, you would type the following command (again, be careful to type that with no carriage returns):
xgrid -job submit /Users/Shared/fasta-tutorial/fasta \
-q /Users/Shared/fasta-tutorial/magic-worm-gene.seq /Users/Shared/fasta-tutorial/chromosomeY.fa
There! You just did it! You sent an xgrid job!! If you look carefully, this is exactly the same command as we typed to start fasta, except it has an additional “xgrid -job submit” in front of it. Those few words “xgrid -job submit” tell xgrid to send whatever command follows to whatever agent is available. The command description is sent from the client to the controller, then to the first agent available, which will run the command exactly as written above. But you don’t have to care about what the controller and the agents are doing. As the client, you simply get a response from the xgrid command that looks like this:
{jobIdentifier = 231; }
Xgrid has acknowledged the submission, has put it in its queue and has given it an identifier that you can use to refer to that job later (‘231’ is just an example; the first identifier you will receive is ‘0’ and that number will go up with each subsequent submission). Before doing anything else with this job, let’s just submit another one immediately, this time scanning chromosomeX:
xgrid -job submit /Users/Shared/fasta-tutorial/fasta \
-q /Users/Shared/fasta-tutorial/magic-worm-gene.seq /Users/Shared/fasta-tutorial/chromosomeX.fa
And we get back the identifier for this job:
{jobIdentifier = 232; }
I encourage you to continue like this with the other 22 chromosomes (you are allowed to use copy and paste!). Pretty soon, you should have your controller quite busy, and probably busy for a while if you have only a few agents. This is where you should realize that Xgrid does actually 2 things: distribute the jobs; queue the jobs. The distribution is about splitting the workload between multiple processors. The queuing part makes sure a new job is started as soon as one finishes (or a new agent is available). While the distinction might seem purely academic, the queuing feature makes Xgrid useful even on a single-machine grid. If you have 200 tasks to run, you probably don’t want to start them all at once and let them compete for the processor resources. Xgrid will instead run them in succession, one after the other. This is the simplest “queuing” you can think of. To take full advantage of your computer, the controller will even decide to run in parallel 2 tasks if it finds that you have 2 processors (maybe your mac will run 8 tasks and use all its 8 cores). In other words, Xgrid can also help to efficiently schedule jobs on a single machine.
As you have just seen, submission is a breeze. Of course, the second part of the process is to retrieve the results. But first, you should check that a job is done. This will be apparent in the Xgrid Admin tool (in the Jobs tab). Or you can simply ask your friend Terminal:
xgrid -job attributes -id 231
which should return something like this:
{
jobAttributes = {
activeCPUPower = 0;
applicationIdentifier = "com.apple.xgrid.cli";
dateNow = 2006-12-08 16:44:16 -0800;
dateStarted = 2006-12-08 12:46:46 -0800;
dateStopped = 2006-12-08 12:47:03 -0800;
dateSubmitted = 2006-12-08 12:42:05 -0800;
jobStatus = Finished;
name = "/Users/Shared/fasta-tutorial/fasta";
percentDone = 100;
taskCount = 1;
undoneTaskCount = 0;
};
}
The important bit here is that jobstatus = Finished. When a job is finished, the agents sends the results back to the controller, and any trace of the job is then removed from the agent. But the controller will keep those results on disk, where they will stay until you specifically ask to delete the job. The results won’t be coming back automatically to the client (the client here is you playing with your friend Terminal). You have to ask the controller for the results. The magic incantation for that is quite simple. Here are 2 examples on how you could do it:
xgrid -job results -id 231
xgrid -job results -id 232 > ~/results-chromosomeX.txt
The first command will directly show you the results of job 231 in the Terminal, which is a bit overwhelming. The second command is more useful, in that it will save the results of job 232 to a file called “results-chromosomeX.txt” in your home folder (also known as “~”). In both cases, you simply provide the job identifier and xgrid knows which job you are talking about. The second command adds a terminal trick that transfers the output to a file instead of the display (this is what the ‘>’ is all about). After you repeat the above for all the chromosomes, you are ready to sift through the results and use your brains for real science again.
Conclusion
In this installment, I hope to have guided you through all the steps for a first-level understanding of Xgrid, starting with the very basic question of when and if you can really use Xgrid for your research, all the way to a first real-world example of how you could use Xgrid to get computations processed faster and automatically on a grid. Fasta makes for a great example of that, in that we were able to split a long computation into smaller pieces that can then easily be sent out on separate processors.
But you may already feel like you have done too many repetitive tasks that the computer should be able to do for you: loading all that data on the agents, submitting all those very similar jobs, retrieving the data, saving it in a convenient location as soon as a job is done. We will explore more of that in the next installements of this tutorial. You will also hear more about the problems you may encounter in maintaining an Xgrid cluster.
Notes
[1] Going back to the paper analogy, writing your manuscript is still going to be more effective if several people work on it: read-proof, format, write a piece of the introduction, write the methods, work on the figures,… However, this requires lots of synchronization and back-and-forth discussion, and can’t be called “embarassingly parallel”. The same is true of computation: when embarrassingly easy parallelization is not possible, there are still possibilities for accelerated computation if things are done in a tightly concerted manner. The code name for that would be MPI, a topic recently covere on MacResearch. I will deliberately ignore this technology in the rest of this tutorial.
[2] Embarrassingly parallel tasks have different granularities. In the case of animation movies, the smallest task you can go down to is the rendering of one frame, which can take several hours, and you can’t divide that task further without significant complications. If you have just one frame to calculate, it is not worth using Xgrid. Fortunately, animation movies need much more than one frame (at least 1440 frames per minute, and many more in you add motion blurring). In the case of a Mandelbrot fractal, you can divide your task as much as you want, because the calculation for each pixel is completely independent of all the other pixels.
[3] Some scientists in that field are interested in large conformational changes, such as those that happen during the folding process, when the protein shape changes radically. These events are “rare”, because they only happen every microsecond or so. Simulations only proceed in picosecond steps, which means it will take on average a billion step to see a change. On one single computer, you may have to run a simulation for decades or centuries before you see that kind of event. However, by randomizing the starting conformation, and running simulations on dozens of thousands on computers in parallel, the probability of these events is increased proportionally to the number of processors, and it becomes feasible to identify these transitions in a time much more compatible with the lifetime of a scientist. This is the idea behind folding@home (note that they don’t use Xgrid!).
[4] Pearson WR, Rapid and sensitive sequence comparison with FASTP and FASTA, Methods Enzymol. 183:63-98, 1990, Entrez Pubmed
[5] For the unix geeks: you could use instead some subdirectory in /usr or /var or some other invisible directory, and make the files world-readable
[6] Actually not the whole chromosomes, just the first 80,000 bases for each of them (only the Y chromosome is provided full-length in the tutorial package). The size of the genome is 3 billion bases and there are 4 possible bases at each position. The human genome would thus use aorund 500 MB, and that would be a little unfair for the MacResearch web servers. You are encouraged to download the full chromosomes, for instance at http://hgdownload.cse.ucsc.edu/goldenPath/hg17/chromosomes/, and run the tutorial with them.
[7] You may be wondering why we install these files on the agents and why we can not let Xgrid install it for us when it needs it. The reason is that Xgrid is not very good at reusing files: it does not do any “caching”. We could let Xgrid package the fasta programs and the human genome together with each job, but the whole thing would have to be uploaded again every time a new job is started. If you want to reuse files for several jobs, it is best to have them pre-installed on each agent, or set up your own cache system (this topic will be explored in a next installement).
[8] Note that the queries run in this tutorial are not something real biologists would normally do. In general, the librairies against which the query is run are not just one big stretch of DNA like a whole human chromosome, but are instead huge lists of genes or of other various piece of DNAs. For instance, the GenBank library contains more than 60 million records, for a total of more than 60 billion bases.
Leave a Reply